
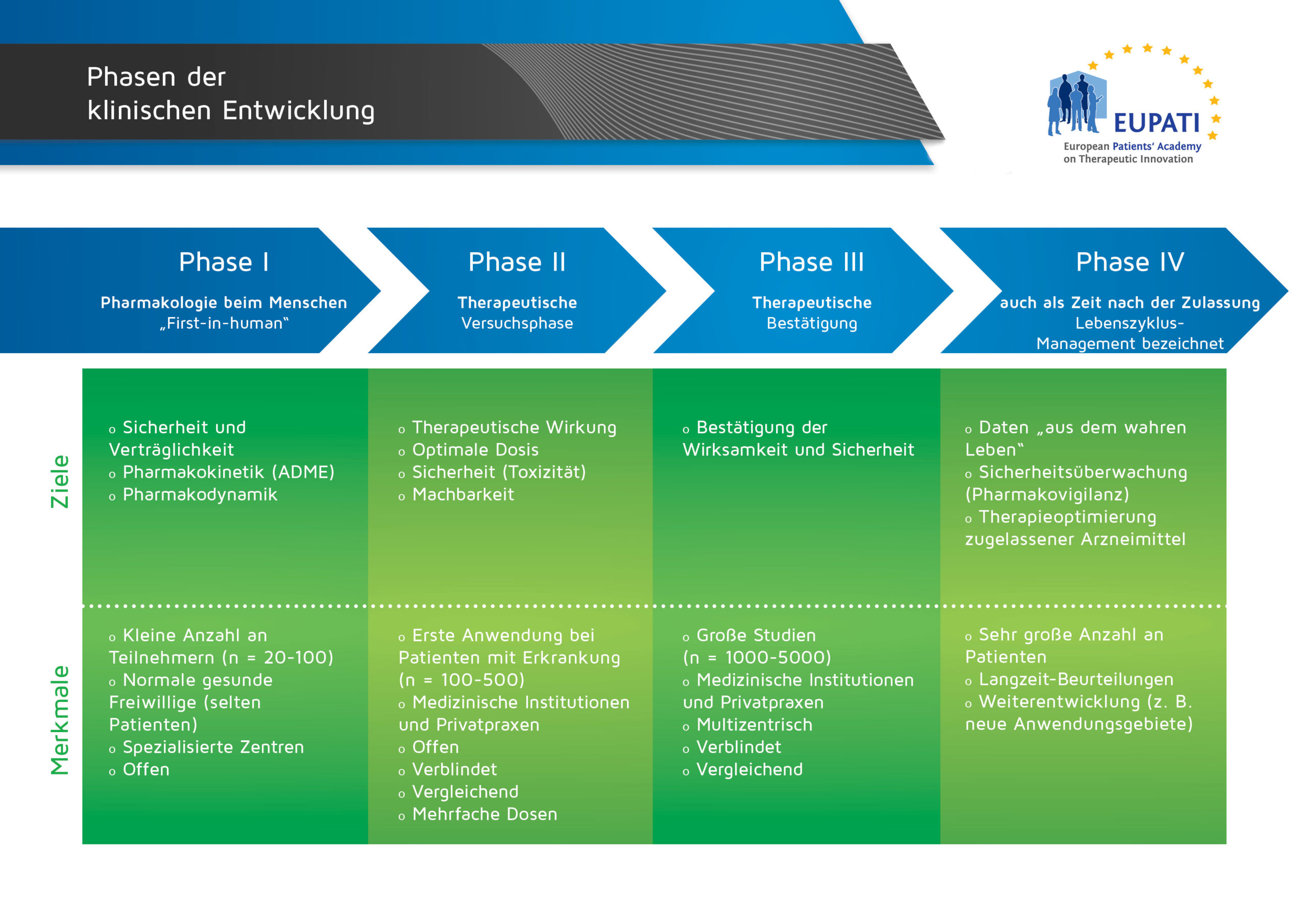
Vor der Markteinführung von Arneimitteln muss jedes Medikament verschiedene Forschungsphasen durchlaufen um die Sicherheit der PatientInnen, die mit dem Medikament behandelt werden, zu gewährleistung und die Wirksamkeit des Medikaments zu bestätigen. Dies ist ein jahrelanger Prozess.
Von First-in-Human bis Real-World-Evidence
Dosisfindung
Dosisoptimierung
Zulassung
Sicherheit
Dosisfindung
10-30 ProbandInnen / national / einige Wochen bis Monate
Eine Phase-I-Studie ist der erste Teil einer klinischen Studie, in der ein neues Arzneimittel am Menschen getestet wird. Diese neue Substanz hat sich im Zell- und/ oder Tiermodell als wirksam erwiesen. Die Teilnehmenden in Phase-I-Studien sind im Allgemeinen gesunde, freiwillige, männliche Personen.
In der Onkologie ist es allerdings häufig eine kleine Gruppe erwachsener PatientInnen mit fortgeschrittener Krebserkrankung, bei der etablierten Standardtherapien keine Wirkung mehr zeigen.
Dies Studien werden in der Regel an spezialisierten Phase-1-Units unter strenger Überwachung der Studienteilnehmenden durchgeführt.
In der Pädiatrischen Onkologie hingegen werden neue Medikamente meistens nicht in Phase I Studien geprüft (Ausnahme bei Erkrankungen, die nur im Kindesalter vorkommen).
In diesen Prüfungen geht es nur darum, die Sicherheit und Verträglichkeit einer neuen Substanz beim gesunden Menschen zu prüfen. Sie dienen hauptsächlich zur Dosisfindung. Zu Beginn werden sehr geringe Mengen des Arzneimittels verabreicht und nur, wenn keine schwerwiegende Nebenwirkungen auftreten, wird die Dosis sukkzesiv erhöht. Ein weiterer Aspekt ist, erste Informationen über die Wirksamkeit zu gewinnen. Dazu werden die so genannten pharmakokinetischen Eigenschaften der Substanz untersucht, das heißt, wie sie sich im menschlichen Körper verteilt, wie sie um- und abgebaut und ausgeschieden wird. Außerdem wird untersucht, ob und wie die Probanden den neuen Wirkstoff vertragen, beziehungsweise ob, welcher Art und in welchem Ausmaß unerwünschte Nebenwirkungen auftreten. Erst wenn alle diese Untersuchungen gezeigt haben, dass der Wirktoff verträglich ist, werden die Untersuchungen an PatientInnen, d.h. an kranken Menschen, fortgesetzt.
Dosisoptimierung
30-150 PatientInnen / häufig weltweit / ca 1 Jahr
In der zweiten Phase einer klinischen Arzneimittelprüfung wird vor allem die Wirksamkeit und die Verträglichkeit einer neuen Substanz untersucht, d.h., ob die neue Substanz die gewünschte positive Wirkung auf eine bestimmte Krankheit beim Patienten zeigt oder nicht und ob und wie das Medikament vertragen wird. Dazu werden ihre so genannten pharmakodynamischen Eigenschaften untersucht, also die Art und Weise, wie ein Arzneimittel das Verhalten eines Zielmoleküls im Körper beeinflusst und die Auswirkungen, die dies auf den Körper hat.
Diese Studien dienen auch dazu, den optimalen Dosisbereich zu bestimmen und zu ermitteln, wie oft das Arzneimittel in welcher Form (z.B. Tablette oder intravenöse Gabe) verabreicht werden soll, um höchste Wirksamkeit und gleichzeitig niedrigstes Risiko für unerwünschte Nebenwirkungen sicherzustellen.
Diese Studienphase wird oft in Phase IIa und Phase IIb unterteilt: Phase-IIa-Studien sind klinische Pilotstudien, die primär die Arzneimittelsicherheit evaluieren, während Phase-IIb-Studien die Wirksamkeit, insbesondere den Dosierungsrahmen und die Sicherheit untersuchen.
Solche Phase-II-Studien schließen in der Regel relativ wenige PatientInnen ein. In der Onkologie handelt es sich hierbei überwiegend um erwachsene Patienten mit bestimmten Krebserkrankungen, die in verschiedenen Studienzentren und häufig auch in verschiedenen Ländern behandelt werden.
Oft wird direkt an die Phase II-Studie eine Phase III-Studie, mitunter zeitlich überlappend, angehängt.
Zulassung
mehrere Hundert bis Tausend Patienten / weltweit / mehrere Jahre
Dritte Phase der klinischen Medikamentenprüfung dient dem Nachweis der Wirksamkeit und Verträglichkeit des Prüfpräparates/neuen Medikaments (bei Zulassungsstudien) oder des neuen Therapiekonzeptes (bei Therapieoptimierungsstudien) im randomisierten Vergleich zur bisherigen Standardtherapie oder einem Placebo. Die Phase III Studie soll an einer größeren PatientInnengruppe bestätigen, was in der Phase II Studie bei einer vergleichsweise kleinen Gruppe festgestellt wurde. Daher wird das Arzneimittel an mehreren tausend Patienten, in der Regel weltweit in randomisierten Prüfungen getestet. Die Studie dauert mehrere Jahre, da die PatientInnen jahrelang nachbeobachtet werden (Follow Up). Ist die Phase III der Arzneimittelprüfung erfolgreich, kann im nächsten Schritt die Zulassung des Medikaments beantragt werden.
Daher werden Phase-III-Studien auch als Zulassungsstudien bezeichnet.
Sicherheit
mehrere Tausend Patienten / weltweit / mehrere Jahre
Phase-IV-Studien erfolgen mit bereits zugelassenen Arzneimitteln.
Ziel dieser Phase ist es, die Wirksamkeit und Sicherheit der neuen Substanz in einer großen PatientInnengruppe sowie in verschiedenen Untergruppen (z.B. verschiedene Alters-und Bevölkerungsgruppen, verschiedene Ethnien, PatientInnen mit verschiedenen Begleiterkrankungen) und unter Alltagsbedingungen zu überwachen. So können hier auch seltene Nebenwirkungen oder Wechselwirkungen mit anderen Medikamenten erfasst werden, die im zeitlich engerem Rahmen der Zulassungsstudie (noch) nicht aufgetreten sind und die Langzeitwirkung oder mögliche Spätfolgen eruiert werden.
Ergebnisse aus diesen Studien führen immer wieder dazu, dass Medikamente Jahre nach der Zulassung vom Markt genommen werden, da die Arzneimittelsicherheit in Hinblick auf Spätfolgen oder Wechselwirkungen nicht mehr gewährleistet ist.


